医疗器械生产企业质量体系考核办法
(2000年5月22日国家药品监督管理局令第22号公布 自2000年7月1日起施行)
第一条 为加强医疗器械管理,强化企业质量控制,保证病患者的人身安全,根据《医疗器械监督管理条例》,制定本办法。
第二条 本办法适用于申请第二类、第三类医疗器械准产注册企业的审查及对企业的定期审查。
下列情况可视同已通过企业质量体系考核:
(一)企业获得国务院药品监督管理部门认可的质量认证机构颁发的GB/T19001和YY/T0287(或GB/T19002和YY/T0288)标准的质量体系认证证书,证书在有效期内的。
(二)已实施工业产品生产许可证的产品,其证书在有效期内的。
(三)已实施产品安全认证,企业持有的产品安全认证证书在有效期内的。
第三条 申请第二、三类医疗器械生产企业质量体系考核,均由所在地省、自治区、直辖市药品监督管理部门受理并组织考核。
国家规定的部分三类医疗器械,由所在地省、自治区、直辖市药品监督管理部门受理后,报国家药品监督管理局,由国家药品监督管理局组织考核。
部分三类医疗器械目录由国家药品监督管理局确定并公布。
质量体系的考核,可委托下一级药品监督管理部门或具有相应资格的第三方机构进行。质量体系考核结果由委托方负责。
第四条 企业在申请产品准产注册前,应填写《医疗器械生产企业质量体系考核申请书》(见附件1),向省级以上药品监督管理部门提出企业质量体系考核申请。
国家规定的部分三类医疗器械的质量体系考核,企业提出质量体系考核申请的同时,向国家药品监督管理局提交被考核产品的《质量保证手册》和《程序文件》。
其它产品的质量体系考核,企业提出质量体系考核申请前,应按《质量体系考核企业自查表》(见附件1的附表)进行自查,填写自查表。自查表填写内容应如实、准确,以备现场考核时查验。
第五条 对二类医疗器械,省、自治区、直辖市药品监督管理部门应对企业填写的《质量体系考核企业自查表》和提供的相关资料进行审核,经审核后签署意见,必要时可对申请企业进行现场查验。
对三类医疗器械,按本办法第三条执行后,质量体系考核申请和考核报告(见附件1、2)应在国家药品监督管理局备案正本(原件)一份。
第六条 考核人员至少应有一人经贯彻GB/T19001和YY/T0287标准的培训,并取得内审员或外审员的资格;考核人员至少由二人组成;确定的考核人员与被考核的企业应无经济利益联系。
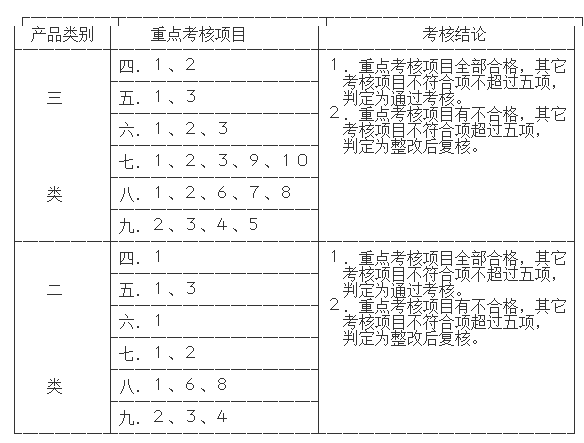
第七条 质量体系现场考核,参照质量体系认证审核的方法;依据附件1自查表确定的内容进行考核,重点考核项目及判定规则为:
考核结论判定为“通过考核”的,对质量体系的评价和存在不合格项要如实陈述,对不合格项给出整改期限。不能如期完成整改的应作为“整改后复核”处理。
第八条 考核结论为“整改后复核”的,以“考核报告”的签署日起,企业必须在半年内完成整改并申请复核,逾期将取消申请准产注册资格。
第九条 企业产品质量体系考核以“考核报告”通过的签署日为准,其有效期为四年;在有效期内企业申请同类产品准产注册,不再进行考核(药品监督管理部门另有规定的除外)。
企业应定期进行质量体系自查,自查结果应按《质量体系考核企业自查表》的规定进行记录、归档。
省、自治区、直辖市药品监督管理部门定期对企业进行体系审查。
第十条 企业通过质量体系考核后,不按规定进行自查、不按质量体系要求组织生产的,经核实,由所在地省、自治区、直辖市药品监督管理部门予以警告,并限期整改。
第十一条 本办法由国家药品监督管理局负责解释。
第十二条 本办法自2000年7月1日起施行。
附表: